一、概述
20世纪80年代,随着色谱技术的高速发展,对化学药品的杂质研究逐渐替代以前只对药品纯度进行控制的理念。BP(1998)在药典中提出了有关物质和残留溶剂的检查通则及杂质研究指导原则。 进入21世纪,随着对杂质研究的逐步深入和分析水平的逐步提高,依靠色谱(HPLC、GC、TLC)、光谱(UV、IR)及联用技术(HPLC-MS、GC-MS)等药品杂质谱控制的理念逐渐形成,2010版中国药典开始尝试对收载的部分化学药品用杂质谱的理念来控制杂质。
众所周知,杂质影响药品安全,尤其是高毒性杂质,所以 CDE 评审中心也重点提出,要对杂质进行充分的研究和控制。但目前药物研发存在的普遍现象是过度解读了“充分研究”这个概念,不管研发的原料药项目工艺是否发生改变,或者与药典或文献收载的原料药的合成路径是否一样;不管工艺中是否会有该杂质的产生或潜在杂质产生的可能性,都先把药典上列出的杂质购买入手或委托合成。这种矫枉过正的现象,已经与我们进行杂质研究的初衷渐行渐远。
二、常用原料药杂质控制涉及的相关法规
1, ICH Q3A:新原料药杂质研究指导原则
2, ICH Q3B:新药制剂杂质研究指导原则
3, ICH Q3C:残留溶剂研究指导原则
4, ICH Q3D:元素杂质研究指导原则
5, ICH M7:基因毒性杂质研究指导原则
6, 2020版中国药典四部 9102 药品杂质分析指导原则
三、原料药杂质的来源及分类
ICHQ3A中对新原料药中的杂质定义为:存在于新原料药中,但是其化学结构与新原料药不一样的任何一种成分。
一般情况下,按照理化性质将杂质分类为:有机杂质、无机杂质及残留溶剂;按照来源,我们可以将杂质分为:工艺杂质(合成过程中未反应完全的反应物及试剂、中间体、副产物等)、降解产物及从反应物或试剂中引入的各类杂质等;从药物毒性出发,又可以将杂质分为基因毒性杂质和普通杂质。
杂质是关乎药品质量安全的关键属性,它贯穿于整个药品的研发过程。因此规范的进行杂质研究,并将其控制在安全、合理的限度范围之内,直接关系到上市药品的质量及安全性。
四、原料药杂质的分析和控制
原料药的杂质谱分析对应于 CTD 格式申报资料的“3.2.S.3.2 杂质情况分析总结”及“3.2.S.4.5 杂质对比研究与杂质谱分析”,当然,CTD 格式中杂质控制体现在 CMC 各个环节,不仅仅局限于某一个模块。
以磷酸奥司他韦为例,结合化合物的结构特点、原料药的合成路线,对其杂质进行分析,浅析原料药杂质的研究过程,包括起始物料、中间体、工艺副产物及降解杂质等,基于 QbD 研究理念,探讨相应的杂质控制策略,以期为仿制药杂质研究工作提供思路。
磷酸奥司他韦,化学名为(3R,4R,5S)-4-乙酰氨基-5-氨基-3-(1-丙氧乙酯)-1-环己烯-1羧酸乙酯磷酸盐,最早是由吉利德开发,2007年,该药物在欧盟批准。用于治疗一岁及以上儿童的季节性和大流行性流感。2009年4月,FDA颁布了奥司他韦的紧急使用授权(EUA),用于治疗和预防甲型H1N1流感。2017年,Chugai公司的3%干糖浆Tamiflu在日本获得批准,用于治疗和预防新生儿和婴儿感染A型或B型流感病毒。
磷酸奥司他韦化合物专利WO1998007685于1997年8月申请,同族专利CN1113053,均届满失效。国内外专利文献查询,磷酸奥司他韦共有6条主要合成路线,作者以Roche第二代路线(参考专利CN1759093A,申请日2004.3.10,预计2024.3.10失效)为例进行分析。具体合成路径如下:
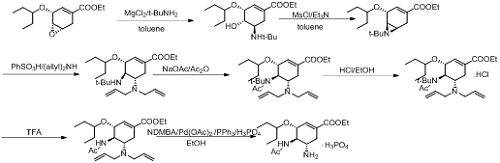
需要指出的是,本文着重对有关物质进行解读,篇幅有限,关于其他如基因毒性杂质、元素杂质、残留溶剂等不作讨论,具体可参照相应指导原则。
4.1 起始物料及引入杂质
以莽草酸为原料先进行一系列反应,最后在碱性条件下成环氧产物。根据其制备工艺,其中市场上售卖的莽草酸基本上为植物(八角、雪松松针、银杏树叶等)中提取出来,其构型单一,均不涉及构型翻转,最后一步关环反应,羟基需从背面进攻,形成环氧化物。从反应机理和条件考虑,产生异构体杂质的可能性很小。结合其制备工艺,其主要有机杂质包括起始物料莽草酸、合成路径中的中间体及其副产物。
尤其需要重点关注部分可引入后续反应的潜在杂质,因为这类杂质与起始物料有相似的结构,可能与起始物料一同进入后续的化反应,而且这类潜在杂质与起始物料的理化性质也较为接近,在后续的工艺步骤对其清除的能力有限。起始物料本身也是成品中的工艺杂质,所以在源头控制和规范好所需要控制的杂质,尤为重要。
基于“以源为始”的杂质研究理念,该起始物料环氧化合物为外购,应严格进行供应商筛选及审计。获得供应商起始物料的生产工艺,结合自身工艺特点,分析起始物料的关键质量属性,特别是起始物料及其杂质在后续工艺中,是否去除或者转化,从而针对性的建立起始物料的内控质量标准。
4.2 中间体及副产物
磷酸奥司他韦的第一步反应为以商业化原材料环氧化合物作为起始物料:
在甲苯溶液中,氯化镁催化下,叔丁胺与环氧化合物进行亲核取代反应,得到化合物A;
再以三乙胺作为缚酸剂,A与甲磺酰氯进行亲核取代反应得到化合物B;
在苯磺酸活化下,化合物B与二烯丙基胺进行亲核取代反应得到化合物C;
以无水乙酸钠钠作为缚酸剂,化合物C与乙酸酐进行乙酰化反应得到化合物D,接着与盐酸乙醇成盐得到化合物E;
在三氟乙酸条件下,化合物E脱去胺基上面的叔丁基得到化合物F;
在无水乙醇、磷酸(85%)和1,3-二甲基巴比妥酸的混合溶液中,醋酸钯催化下,化合物F脱去烯丙基,然后成磷酸盐得到磷酸奥司他韦粗品;
磷酸奥司他韦粗品在无水乙醇和纯化水中重结晶得到磷酸奥司他韦纯品。
对于中间体和副产物等杂质,应充分体现过程控制的理念。
由于磷酸奥司他韦的合成路线比较长,如果把评估的中间体及潜在工艺杂质,都放到成品里面进行控制,就会显得很繁冗且提高最后成品方法开发的难度。因此在每步反应时进行过程控制,将每步的中间体纯度和反应物的转化率控制在合理的限度水平,才能保证成品的质量。对各步骤可能产生的副产物,需要跟踪其在后续工艺中的杂质去向,并且根据多批次累积的数据,合理制定各步反应的中控及中间体标准。
以磷酸奥司他韦第三、四步反应为例,化合物B经苯磺酸催化,加热条件下,与二烯丙基胺进行亲核取代反应得到化合物C;以无水乙酸钠钠作为缚酸剂,化合物C与乙酸酐进行乙酰化反应得到化合物D。起始物料与化合物B有相似的结构,在第三步反应过程中,环氧闭环会打开,与二烯丙基胺发生亲核取代反应,产生新的工艺杂质a,反应如下:
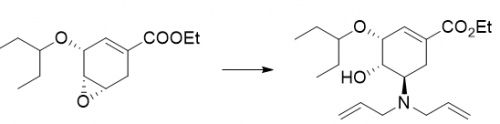
工艺杂质a可继续参与后续的乙酰化等反应。因此,为了减少下一步反应产生各种副产物,可以在上一步把可能参与下步反应的潜在杂质通过溶剂溶解差异性等方法清除或者控制在一个合理的限度内,从而达到不影响最终产品的目的。
4.3 降解杂质
起始物料、中间体及最终成品如果因为储存方式不当,都会产生降解杂质。我们可以根据稳定性试验指导原则,设计不同的强降解试验,从而在短时间内获得大量降解杂质的信息。
ICH Q1A 中明确指出了强降解试验的内容,包含了高温、氧化、酸碱破坏、光照等破坏方式,试验样品可以以原料药本身的固体状态进行破坏,也可以以溶液的方式进行破坏。通常情况下,通过不同的试验条件筛选出合适的强降解条件,主成分降解 10% 左右。对于在剧烈试验条件下仍然稳定的化合物,没有必要一定要采取某种方式使其降解。
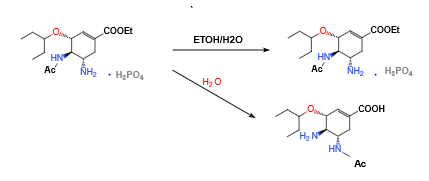
USP43 收载了磷酸奥司他韦两个杂质,其中杂质A化学名为(3S,4R,5S)-4-乙酰氨基-5-氨基-2-叠氮基-3-(戊-3-基氧基)环己烷甲酸乙酯,就是由磷酸奥司他韦原料药在用乙醇精制过程中水解所产生的降解杂质。将磷酸奥司他韦原料药在 100℃ 下进行水解反应,发现原料药大量水解,产生杂质A。
4.4 有机试剂及其转化物
磷酸奥司他韦合成过程中使用了多种有机溶剂或试剂,第一步反应中,在甲苯溶液中,氯化镁催化下,叔丁胺与起始物料中环氧进行亲核取代反应;第二步反应中,三乙胺作为缚酸剂,化合物A与甲磺酰氯进行亲核取代反应;第三步反应中,在苯磺酸活化下,化合物B与二烯丙基胺进行亲核取代反应;第四部反应,无水乙酸钠钠作为缚酸剂,化合物C与乙酸酐进行乙酰化反应得到化合物D等等,上述反应中的有机试剂如叔丁胺、甲基磺酰氯(容易水解,产生基因毒杂质,此文不作赘述)、乙酸酐、1,3-二甲基巴比妥酸等都不仅仅作为溶剂,都参与了反应。应该严格跟踪这些有机试剂及其转化产物的去向并进行研究分析,多批次积累数据,制订合理的内控标准。
五、总结与讨论
有机杂质是原料药质量研究过程中的重点和难点,不管是新药还是仿制药,杂质的研究贯穿整个研发始终。微谱在有机杂质原料药方面有丰富经验,随着对质量控制理念认知的不断变化和对药品安全的高度关注,我们研究杂质应当以实际工艺路线为基础,参考各国药典、文献,合理且充分地研究对原料药质量产生影响的各类杂质,保证源头控制、过程控制、终点控制,每一步有的放矢,从而实现药品的安全、可靠性。
免责声明:市场有风险,选择需谨慎!此文仅供参考,不作买卖依据。